

Maria Kontoyianni, PhD

Professor of Pharmaceutical Sciences
Phone: 618-650-5166
Fax: 618-650-5145
Email: mkontoy@siue.edu
B.S., Pharmacy, University of Athens, Greece
Ph.D., Medicinal Chemistry/Computational Chemistry, University of North Carolina, Chapel Hill
Post-doctoral, Biomedical Engineering, University of Washington, Seattle
Biography
Dr. Kontoyianni holds a PhD in computational chemistry from the University of North Carolina, Chapel Hill, where she worked under the supervision of Professor Phil Bowen. After a post-doctoral fellowship with Professor Terry Lybrand at the University of Washington, she joined ZymoGenetics, where she used computational methods in support of osteoporosis. She then moved to Research & Development of Fortune 500 companies such as Johnson & Johnson and Procter & Gamble, applying structure-based design to various therapeutic targets ranging from hit to lead optimization. In her most recent post and prior to joining academia, she was the Head of Drug Discovery at Crystax, a fragment-based biotechnology firm in Barcelona. She sits on the editorial board of several journals, holds nine patents, is a clinical consultant, panelist for the European Commission large scale grant application proposals, has been an NIH ad hoc reviewer for Bioengineering and Small Business and Technology Transfer in Drug Discovery and Development, and is regularly invited to speak at Drug Discovery conferences and symposia. Her area of expertise includes virtual screening, structure- and ligand-based molecular design, and hybrid computational approaches toward drug discovery.
Research and Representative Publications
Dr. Kontoyianni’s laboratory focuses on:
- Drug discovery approaches to specific targets or disease areas. A major area in the laboratory involves ligands and their involvement in modulating protein properties for disease-related targets, e.g., somatostatin, cytochrome P450s, and toll-like receptor four.
Somatostatin:
- To rationalize observed affinities of internal compound series in the somatostatin subtype-4 receptor (sst4), we are pursuring a structure-based design strategy. Toward that end, we construct homology models (model building) and dock the active leads into the model-built sst4 structures, as well as in the recently released cryo-EM sst4. Comparative results have enabled us to explain why small differences in structures result in significant at times effects on affinity. Further, in order to prioritize future synthetic targets, we have explored the relationship between chemical structure and affinity with a global QSAR model, along with graphical projections onto chemical functionalities.
Liu, Z.; Crider, A.M.; Ansbro; D., Hayes, C.; Kontoyianni, M. A Structure-Based Approach to Understanding Somatostatin Receptor-4 Agonism (sst4), J. Chem. Inf. Model. 2012, 52, 171-186
Neumann, W.L.; Sandoval, K.E.; Mobayen, S.; Minaeian, M.; Kukielski, S.G.; Srabony, K.N.; Frare, R.; Slater, O.; Farr, S.A.; Niehoff, M.L.; Hospital, A.; Kontoyianni, M.; Crider, A.M.; and Witt, K.A. Synthesis and Structure-Activity Relationships of 3,4,5-Trisubstituted-1,2,4-Triazoles: High Affinity and Selectivity Somatostatin Receptor-4 Agonists for Alzheimer’s Disease Treatment, RSC Med. Chem. 2021, 12, 1352-1365
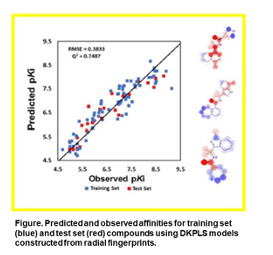 Slater, O. and Kontoyianni, M. A Computational Study of Somatostatin Subtype-4 Receptor Agonist Binding, SN Applied Sciences 2022, 4, 140.
Slater, O. and Kontoyianni, M. A Computational Study of Somatostatin Subtype-4 Receptor Agonist Binding, SN Applied Sciences 2022, 4, 140.
Crider, A.M.; Hospital, A.; Sandoval, K.E.; Neumann, W.L.; Garic, L.; Ingold, K.; Dunahoo, M.; Srabony, K.N.; Frare, R.; Slater, O.; Peel, N.; Kontoyianni, M.; Witt, K.A. 3-Thio-3,4,5-Trisubstituted-1,2,4-Triazoles: High Affinity Somatostatin Receptor-4 Agonist Synthesis and Structure-Activity Relationships, RSC Med. Chem., 2025, 16, 945-960.
- To explore the significance of sst4 macromolecular flexibility, we have been carrying out molecular dynamics simulations on agonist/sst4 complexes.
Kontoyianni, M. and Grossfield, A. Molecular Dynamics Simulations Reveal Interactions of Designed Agonists with Somatostatin Subtype-4 Receptor, J. Comput. Aided Mol. Des. 2025 (Submitted).
- To identify parameters that correlate best with affinity, Quantum Mechanics/Molecular Mechanics (QM/MM) simulations are used. Consideration is given to pair-wise comparisons or matched molecular pairs with focus on the effects of chemical substitutions on ligand properties and energetics. Amino acid interactions consisted among all refined agoanist-bound complexes are identified, as well as those that differentiate high affinity binders.
Slater, O. and Kontoyianni, M. Elucidating Structural and Molecular Requirements of Somatostatin Subtype-4 Agonist Bound Complexes Using Quantum Mechanics Approaches, Org. Biomol. Chem., 2025 (Submitted).
CYP3A4:
- To map the binding pocket of substrates and inhibitors of the isoform which metabolizes over one-third of drugs, CYP3A4, we have employed ensemble-docking of a 195-substrate library using induced fit and GOLD docking algorithms and a number of scoring functions. Our analyses suggested that certain residues make favorable interactions with bound substrates and that multiple receptor conformations enhance the accuracy of catalytic prediction. We carry out multiple MD simulations to quantify the CYP3A4 channels, and characterize the behavior of gating residues. Our findings shed light on the equilibrium behavior of these residues and channels in the unbound state.
Hayes, C.; Ansbro, D.; Kontoyianni, M. Elucidating Substrate Promiscuity in the Human Cytochrome 3A4, J. Chem. Inf. Model. 2014, 54, 857-869.
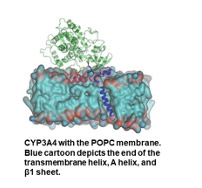
Kontoyianni, M. and Lacy, B. Toward Computational Understanding of Molecular Recognition in the Human Metabolizing Cytochrome P450s, Curr. Med. Chem. 2018, 25, 3353-3373
Ackad, E.; Biggers, L.; Meister, M.; Kontoyianni, M. Equilibrium Landscape of Ingress/Egress Channels and Gating Residues of the Cytochrome P450 3A4, PLoS ONE 2024, 19(3): e0298424.
Toll-like Receptor 4 (TLR4):
- In collaboration with UMSL, SLU and SIU Medical School, we are working on an integrated approach to chemotherapy and inflammation. Toll-like receptors (TLRs) and their accessory proteins are an integral component of the human innate immune response. Overactivation of the TLR system can play a deleterious role in both acute (sepsis, septic shock) and chronic (Alzheimer’s disease) disease processes. Lipid A, the active component of LPS, binds pathogen recognition receptors such as the Toll-like receptor 4 (TLR4), myeloid-differentiation factor 2 (MD2), and cluster of differentiation 14 (CD14), ultimately leading to proinflammation. We aim at studying and disrupting LPS/TLR4/MD2 and pfAβ/CD14/TLR interactions with probes and ultimately drug-like alternatives to phospholipids.
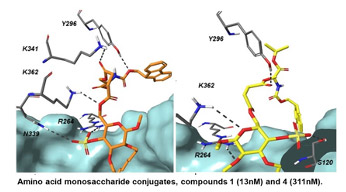
Slater, O.; Miller, B; Kontoyianni, M. Decoding Protein-protein Interactions: An Overview, Curr. Topics Med. Chem. 2020, 20, 855-882.
Slater, O.; Dhami, K.; Shrestha, G.; Kontoyianni, M.; Nichols, M.; Demchenko, A. The Development of a Simple and Effective Lipid-A Antagonist Based on Computational Prediction, ACS Inf. Dis. 2022, 8, 1171-1178.
2. Development of Computational Methodologies and Classification Models. A major area in the laboratory involves the investigation of protein binding sites and requirements for binding. We correlate the nature of binding pockets with small molecule variables to predict potential for drug-protein associations, and also navigate through protein families to predict putative protein function. Another thrust in the laboratory involves virtual screening (VS), its performance and limitations. We are carrying out multiple VS experiments to determine how library size and homology model template differences affect active compound retrieval. We also consider the role of plasticity of macromolecules on VS’s accuracy at retrieving a high fraction of the actives seeded in prepared libraries.
Kontoyianni, M. and Rosnick, C. Functional Prediction of Binding Pockets,” J. Chem. Inf. Model. 2012, 52, 824-833.
Kontoyianni, M. Docking and Virtual Screening in Drug Discovery, In Proteomics for Drug Discovery, book in the series of Methods in Molecular Biology, Iulia M. Lazar, Maria Kontoyianni, and Alexandru C. Lazar, (Eds.); Springer, Heidelberg, 2017, pp 255-266.
Slater, O.; Kontoyianni, M. The Compromise of Virtual Screening and its Impact on Drug Discovery, Expert Opin. Drug Discov. 2019, 14, 619-637.
Kontoyianni, M. Library size in virtual screening: is it truly a number’s game? Expert Opin. Drug Discov., 2022, 17, 1177-1179.
Kontoyianni, M. Structure-Based Virtual Screening: Theory, Challenges and Guidelines.” In: Comprehensive Pharmacology; Elsevier, 2022, vol. 2, pp. 539–552. Elsevier.





